








Gait and Balance Webinar
Download the Video
LINK
What is Ataxia?
Ataxia means "imbalanced" or "unsteady", and it refers to one's gait. One who is intoxicated is said to have "temporary ataxia". Ataxia is a symptom of a degenerative cerebellum but it has come to be the name of the disease. The first visible symptom of the disease is ataxia, that is, staggering or unsteady of gait.
The cerebellum is that part of the brain located at the base of the skull, at the back at the top of the spinal cord. 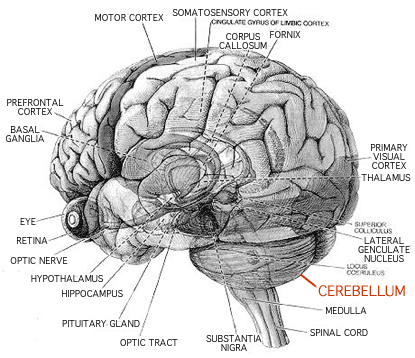 Besides having this affliction in general, it manifests itself particularly, in MSA - Multiple System Atrophy. This means a gradual shutting down of the organs of the body. The urinary (bladder) system has closed down, so that I require a permanent catheter. I have one inserted through the abdominal wall. It is called a suprapubic catheter. The next thing to go is the bowel. It has already shown signs of deterioration. I suppose I'll have to be confined to a nursing home when that is in full bloom! I will be in a wheelchair by then, unable to walk.
Besides having this affliction in general, it manifests itself particularly, in MSA - Multiple System Atrophy. This means a gradual shutting down of the organs of the body. The urinary (bladder) system has closed down, so that I require a permanent catheter. I have one inserted through the abdominal wall. It is called a suprapubic catheter. The next thing to go is the bowel. It has already shown signs of deterioration. I suppose I'll have to be confined to a nursing home when that is in full bloom! I will be in a wheelchair by then, unable to walk.
Causes of Ataxia
"Most often ataxia is caused by loss of function in the part of the brain which serves as the 'coordination centre', which is the cerebellum."
"…The right side of the cerebellum controls coordination on the right side of the body, and the left side controls coordination on the left. The central part of the cerebellum is involved in coordinating the very complex movements of gait, or walking. Other parts of the cerebellum help to coordinate eye movements, speech and swallowing.”
"… Ataxia may be caused by the dysfunction of the pathways into and out of the cerebellum."
"… Information comes into the cerebellum from the spinal cord and other parts of the brain and signals from the cerebellum go out to the spinal cord and to the brain. Although the cerebellum does not directly control strength … or feeling …, the motor and sensory pathways must work properly to provide the correct input into the cerebellum."*
*"Frequently Asked Questions about the CLASSIFICATION OF ATAXIA", National Ataxia Foundation, 2600 Fernbrook Lane, Suite 119, MPLS, MN 55447. www.ataxia.org
Kinds of Ataxia
There are two big categories of ATAXIA:
The Inherited ATAXIA is divided into Dominant and Recessive.
Dominant is divided as follows:
Most types of SCA (Spinocerebellar Ataxia) are inherited in an autosomal dominant pattern. There are currently 29 different ataxias. They are numbered in the order they were found. DRPLA (Dento-rubro-pallido-luysian atrophy) is also a dominant form of ataxia. When a parent with dominant ataxia produce offspring, each of the offspring will either inherit ataxia or will not inherit ataxia depending on which genes are passed on by the parent.
Recessive:
Initially, ataxia as a disease was categorized in two ways. If ataxia symptoms developed at an early age you, had Friedreich's ataxia. If ataxia symptoms enveloped after age 20, it was given the name Maria's ataxia.
Friedreich's Ataxia was named after Nicholaus Friedreich, a doctor in Germany, who first diagnosed a form of ataxia that was inherited, in the early 1860's.
Friedreich’s ataxia is an inherited condition, due to a mutation on chromosome 9. This results in degeneration of nervous tissue in the spinal cord and in the nerves connecting to the spinal cord with muscles, joints, and skin (peripheral nerves). It results in an inability to coordinate voluntary movements of the head, neck, arms, and legs. Those affected may exhibit shakiness and instability of movement, which is typical of this disease.
Although rare, FRDA has an incidence of 1 in 30,000-50,000. Both parents must be carriers of the diseased gene in order to have an affected child; if both parents are disease gene carriers, the chance that any child of theirs will have a “double dose” of the disease gene and develop the disease is 25 percent.
SCA1 (Spinocerebellar Ataxia Type 1)[Schut's, hereditary olivopontocerebellar atrophy, or Marie's]: the gene was identified in 1993; onset - mid-thirties, incoordination of hands and ataxia, swallowing and speech affected; some develop neuropathy. Spasticity, weakness and/or memory problems. It is autosomomal dominant. Children of a parent with SCA1 are 50% likely to have the disease. It is passed directly from parent to child, regardless of sex of carrier parent or of the child inheritor.
SCA2 (Spinocerebellar Ataxia Type 2) This type is linked to chromosome 12 and is autosomal dominant, on-set in adulthood. There is often, as symptom, neuropathy, slow eye movement and ataxia. The gene was discovered in 1996.
SCA3/MJD: (Machado-Joseph Disease, SCA3)The gene was discovered in 1993. The onset is mid-adulthood. The symptoms are ataxia, incoordination, spasicity, rigidity and loss of muscle bulk. It is also autosomal dominant.
SCA4(Spinocebellar Ataxia type 4) Autosomal dominant, On-set in adulthood. The symptoms are ataxia, slowness and neuropathy. It is located on chromosome 16, but the gene has not been found yet.
SCA5(Spinocerebellar Ataxia Type 5)[Holmes or Lincoln's Ataxia]. The on-set is after fifty years of age. The gene is not yet identified, but has been localized to chromosome 11 and is autosomonal dominant. Symptoms are generally confined to the cerebellum; hands, arms, legs and speech and ataxia. Usually thinking, swallowing, bowel, bladder and speech are not affected. Lincoln did not have the affliction himself, but a part of his family did.
SCA6 (Spinocerebellar Ataxia Type 6) Was recognized as a distinct form of ataxia in 1997. There are eye problems - "nystagamous"- connected to it, momentary imbalance which worsens over time and dizziness when moving or turning. Some have a mild loss of sensation in the feet and legs.
SCA7 (Spinocebellar Ataxia Type 7) [autosomal dominant cerebellar ataxia type 2 or ataxia with pigmentary retinopathy] The on-set is ordinarily in the mid-twenties, but sometimes it is as early as four years of age. The gene has not yet been identified, but is known to be located on chromosome 3. The earliest symptoms have to do with vision; also ataxia, slow eye movements, or mild changes in sensation.
SCA8 (Spinocerebellar Ataxia Type 8) is an autosomal dominant cerebeller ataxia. The gene was found in 1999. The genetic aspects of SCA8 are somewhat unusual, and researchers are working to determine exactly how changes in the gene correspond to symptoms in SCA8 families. Slow progression limb and gait ataxia and slow hesitant speech may be early symptoms.
SCA10 (Spinocerebellar Ataxia Type10) is characterized by ataxia and ataxia seizures. The responsible gene was identified in 2000.
SCA11 (Spinocerebellar Ataxia Type11) Adult onset, very slowly progressive form of pure cerebellar ataxia. It has been reported in a single family in England.
SCA12 (Spinocerebellar Ataxia Type12) adult onset described in several families of European decent. Symptoms usually appear in the 30’s with a tremor of the head or arms. May also have hyperactive reflexes, slurred speech and cerebral atrophy.
SCA13 (Spinocerebellar Ataxia Type13) Affected individuals have a slowly progressive ataxia beginning in childhood and also have a mild retardation. Very rare type of ataxia.
SCA14 (Spinocerebellar Ataxia Type14) Was initially described in a single family in Japan in 2000; affected individuals were accompanied by twitchy movements of the body called myoclonus.
SCA15 (Spinocerebellar Ataxia Type 15) Slow progressive form of pure cerebellar ataxia, reported in a single family in Australia.
SCA16 (Spinocerebellar Ataxia Type 16) Adult onset, pure cerebellar ataxia. Described in one Japanese family. People in this family have ataxia, dysarthria (unclear speech), and one third also have head tremor.
SCA17 (Spinocerebellar Ataxia Type 17) Reported in one family of Belgian heritage. Affected family members had the onset of ataxia had dementia in their 30’s or 40’s, Very rare.
Other ataxia’s not listed here are very rare
DRPLA (Dentato-rubro-pallido-luysian atrophy) a rare condition, named after the parts of the brain most affected by the disease. The gene responsible for this type was identified in 1994. It is from a group called after a particular kind of gene change called a "trinucleotide repeat expansion".
Ataxia may not be noticed because other symptoms are more in evidence. Hands are affected and arms move involuntarily, showing tremour, spasicity and rigidity. If on-set is in childhood or adolescence, epilepsy may be present. It is an autosomal dominant disease.
EA1 (Episodic ataxia-1).The on-set of this type is usually during the teenage years. The gene was discovered in 1994. The episodes of EA1cannot are controlled with acetazolamide. It is also an autosomal dominant disorder. It is known to be located on chromosome 19.
EA2 (Episodic ataxia-2).Located on chromosome 19, but its gene has not yet been discovered. As with EA1, it is an autosomal dominant disorder. Its on-set is chiefly during teenage years. It has episodes of ataxia and garbled speech, usually brought on by exercise.*** Unlike EA1, its episodes are of longer duration - sometimes hours - with nystagamus, or rapid eye movements). The medication acetazolamide helps sometimes with no.2 but not no.1.
Ataxia telangiectasia (AT) Neurological symptoms appear early in life, with ataxia appearing when the child begins walking. Some children have mental retardation, with slowness of movement, drooling and dystonia (poor muscle tone). Some may manifest oculomotor apraxia (muscular inability, as it applies to the eyes). Peripheral neuropathy may develop as well as trouble with speech and swallowing.
There is a susceptibility to infection and different types of cancer. If more than one sibling has this ailment, then an autosomal recessive disorder is likely. Aggressive therapies can improve symptoms. If one is a carrier of AT, and not a sufferer of it, he should consult a doctor about his susceptibility to other diseases.
Other Kinds of Ataxia:
Structural Disorder: In this category are Joubert's syndrome, Gillespie's syndrome, granule cell hypoplasia and another number of rare conditions. These are rare and are autosomal recessive. This begins in the womb.
Metabolic Disorders: Caused by enzyme deficiencies and are autosomal disorders. Toxins, which the enzymes are supposed to break down, cause the syndrome, and proteins which the enzymes are supposed to create. These have the symptom of ataxia. These ought to be examined because the symptoms can be controlled.
Degenerative Disorders: of unknown origin can cause ataxia as a main feature. These disorders are very rare.
For more information, go to the National Ataxia Foundation: www.ataxia.org or call at (763) 553-0020.
**CAVEAT (Warning): I am not a medical person, much less a doctor. Everything I know about this disease, or affliction, I learned from books and the internet, and even then I may be in error! If you suspect you have anything such as I have described, DISCUSS IT WITH YOUR DOCTOR. Do not depend entirely on what you read here.